|
Yoichi Yamaguchi, Yasunori Yokomichi, Shiyoshi Yokoyama and Shinro Mashiko Molecular simulations of the four generations of novel azobenzene dendrimers were conducted by using molecular dynamics (MD) and molecular orbital (MO) methods. The molecular structures of these dendrimers in chloroform solution and a model of Langmuir-Blodgett (LB) film containing the gas phase and amorphous solid states were decided by using a MD algorithm. From the MD-optimized geometries of the dendrimers, the visible absorption spectra and second-order molecular hyperpolarizabilities ( 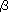 ) were calculated by using the semiempirical MO method, CNDO/S-CI level of theory. It is found that the dendrimers in chloroform solution have favorable rod-shaped structures giving values larger than those of the dendrimers in the gas phase. The simulations give values (for incident light with ) were calculated by using the semiempirical MO method, CNDO/S-CI level of theory. It is found that the dendrimers in chloroform solution have favorable rod-shaped structures giving values larger than those of the dendrimers in the gas phase. The simulations give values (for incident light with  = 1064 nm) for the second, third, and fourth generations in chloroform solution that are respectively 2.0, 6.5, and 9.1 times the value for the first one (75 = 1064 nm) for the second, third, and fourth generations in chloroform solution that are respectively 2.0, 6.5, and 9.1 times the value for the first one (75  10-36 esu). The tendency of these values is in agreement with that of values obtained experimentally. It is assumed that the value of the azobenzene units in the LB films are smaller than those of the azobenzene units in chloroform because the units in the film have staggered conformations. 10-36 esu). The tendency of these values is in agreement with that of values obtained experimentally. It is assumed that the value of the azobenzene units in the LB films are smaller than those of the azobenzene units in chloroform because the units in the film have staggered conformations.
|